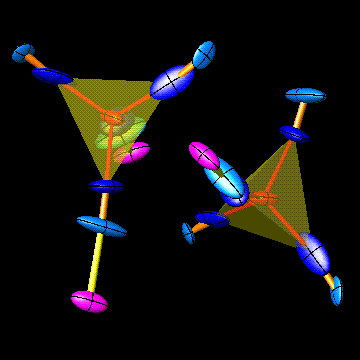 *
* Shape Software
*
ATOMS
for atomic-structure display
*
SHAPE
for crystal morphology
*
VIBRATZ
for normal-coordinate vibrational analysis
*
CRYSCON
crystallographic conversion and utility
*
*
Software for Windows, Macintosh and Linux
*
*
Shape Software
*
ATOMS
for atomic-structure display
*
SHAPE
for crystal morphology
*
VIBRATZ
for normal-coordinate vibrational analysis
*
CRYSCON
crystallographic conversion and utility
*
*
Software for Windows, Macintosh and Linux
*
Contents
ATOMS
description
ATOMS images
SHAPE description
SHAPE images
VRML (3D) Files
VIBRATZ description
CRYSCON description
File
Formats Supported
Prices and Ordering Information
Update on 3D Stereoscopic Viewing
Technology for stereoscopic "true" 3D viewing with moderately-priced hardware has advanced rapidly. The ATOMS V6.5 and SHAPE V7.4 releases for Windows support many of the new displays through quad-buffered OpenGL and Direct3D. As of June 2011 ATOMS and SHAPE support Stereo viewing with Direct3D in a window as well as fullscreen. See the Stereo Viewing page for more details.
Movie Files.
The ATOMS V6.5 and SHAPE V7.4 releases have the ability to write video or movie files of rotation or vibration. These can be written in the platform-independent Flash (.swf) format or as .avi files which can be converted to .mp4, .webm or .ogg files for native play in browsers (files shown here are .mp4)
See a movie of thermal motion in corundum.
With release of the Catalina operating system (OS 10.15) support for 32-bit programs on Macintosh has been discontinued. All Shape Software programs for Macintosh are 32-bit and there are no plans to update to 64-bit, which would require extensive rewriting. In order to run these programs in a Macintosh it will be necessary to operate an older OS, such as Mojave (OS 10.14). There are several ways to do this on a single computer, as discussed here. Current Mac users may also be eligible for free or low-cost switch to the Windows version. Contact support@shapesoftware.com.
ATOMS for
atomic-structure display 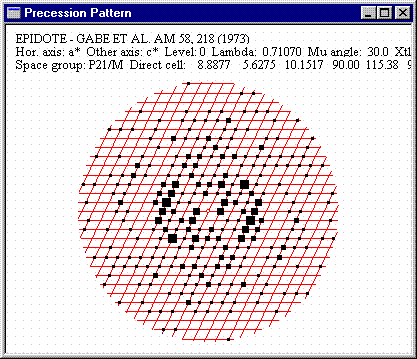
ATOMS is a program for drawing all types of atomic structures, including crystals, polymers and molecules. It can make fully "three-dimensional" color drawings using the latest system software, or it can make simple schematic black-and-white drawings for reproduction on a small scale in publications - or virtually anything between these extremes.
The representation can be ball-and-stick, wire-frame, interpenetrating atoms (space-filling), coordination polyhedra, or combinations thereof. A separate thermal ellipsoid mode draws atoms and bonds in the style of ORTEP. Solid ellipsoids can also be shown in the 3D mode. Drawings can be orthographic or perspective, and stereopairs can be automatically drawn. Anaglyphs (two-color superimposed stereopairs) can also be drawn in wire-frame representation. Atoms and bonds can be labeled automatically. Atomic vectors can show magnetic spins (using Shubnikov symmetry if desired) or any other vectorial properties, and may also be used to show atomic displacements in vibrational modes (using output from VIBRATZ - see below). A library of 13 basic inorganic crystals is now included at no charge, along with other sample structures.
ATOMS will use lattice translations and space-group symmetry
to produce as
many generated atoms as desired for a crystal or polymer, using any of
several
boundary options: 1) unit cell; 2) chosen crystal forms; 3)
translations
(unit-cell) limits; 4) slice (growth layer); 5) sphere; or 6) isolated
molecules.
Bonds and polyhedra are located automatically, from minimal
specifications.
After generation of all atoms, it is possible to move or otherwise
modify
individual atoms. Molecules can be isolated in crystal structures and
multiple
structure fragments can be rotated and translated
independently. 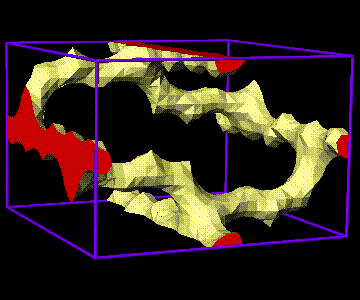
There are many options for rotation in real-time and analytically, including alignment on crystal vectors and/or faces. The drawing can be scaled automatically for a specified frame, or in absolute units.
ATOMS will import several kinds of atomic-coordinate files (see File Formats Supported). A free-form input option makes it easy to read most other text files containing atomic coordinates.
Printed and file output is made at the maximum resolution of the printer or other device, scaled automatically to the specified frame if desired. Output to a PostScript printer can be made directly in the PostScript language for maximum speed and resolution. Windows and Mac versions of ATOMS will both write Encapsulated PostScript, VRML (3D), PNG raster and HPGL files. The Windows version will write Windows metafiles (enhanced), and .BMP, .PCX, .TIF or .PNG raster files. The Macintosh version will write PICT files as either Pictures (metafiles)or bitmaps and .PNG raster files. All output may be either color or black-and-white. Colors for color display and output are separate from gray shades for black-and-white output.
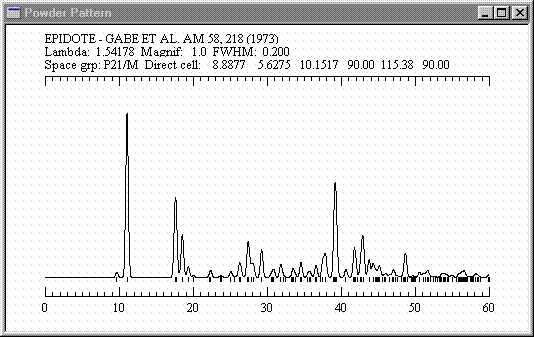
New for Version 5.0. The new "Cavities" display mode shows open space in crystals - see the drawing above (zeolite ZSM5). You can display or print simulated powder diffractograms or precession photographs, or print out the structure factors and powder intensities. Hachure patterns for polyhedra and atoms improve the ability to differentiate types in black-and-white drawings. There are many user interface improvements, including rotation with the mouse.
New for Version 5.1. Over 20 additions and extensions from V5.0, including layer expansion (e.g. for modeling clay minerals), new options for locating molecules, and extensions to both 2D and 3D drawing.
New for Versions 6.0 and 6.1. Additions include rotation movies, POV-Ray files, magnetic wave vector simulation, randomized atom properties for simulating solid solutions, new atom deletion tool, general planes and cylinders in 3D mode and z-matrix input for generation of molecules.
New for Versions 6.2 and 6.3. Additions include half-and-half bond coloring, movable labels, hydrogen atom location, animation of vibrational modes and thermal motion in VIBRATZ files and others.
New for Version 6.4. Now supports 3D stereoscopic viewing with either OpenGL quad-buffering or Microsoft Direct3D (DirectX).
New for Version 6.5. Supports writing files - StereoLithograph (.stl) and Wavefront (.obj) files - for direct output for 3D printing.
ATOMS for Windows V6.5. This is a 32-bit program which will run on most versions of Windows from XP on.
ATOMS for Macintosh V6.5. For Intel on OS 10.4 or later.
ATOMS for Linux/GTK V6.5. For Intel x86.
Library of over 220 Minerals. This library includes the data files used for the structure illustrations in the 8th edition of Dana's System of Mineralogy (Dana's New Mineralogy - John Wiley).
Add ATOMS to your Shopping Cart
View some ATOMS images in raster format (may take
some time to download).
Color quality in 3D files is less than ideal because of the necessity
to keep
file size small.
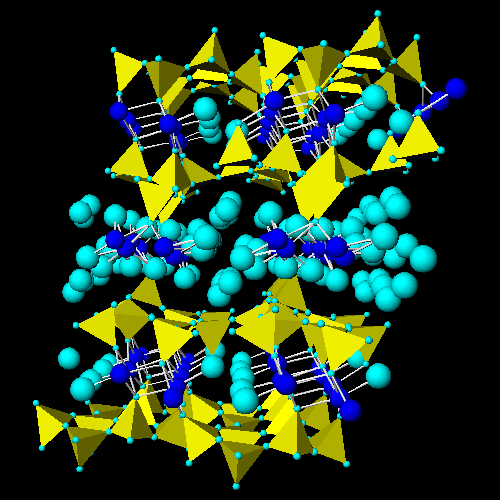
![]() Carnallite.
The full image is 32K
Carnallite.
The full image is 32K
![]() MnO (FCC)
magnetic structure (3D). The full image is 16K
MnO (FCC)
magnetic structure (3D). The full image is 16K
![]() Garnet. The
full image is 32K
Garnet. The
full image is 32K
![]() Okenite. The
full image is 35K
Okenite. The
full image is 35K
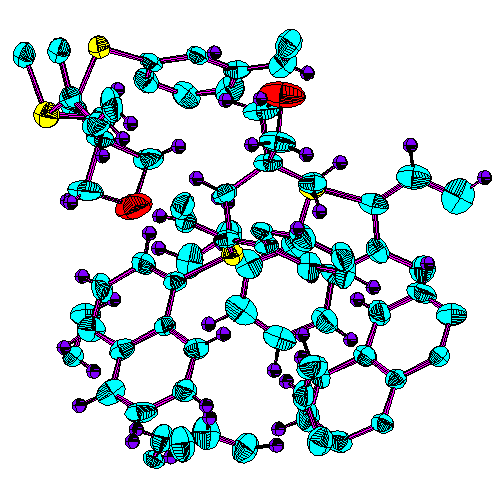
![]() ORTEP drawing.
The full image is 16K
ORTEP drawing.
The full image is 16K
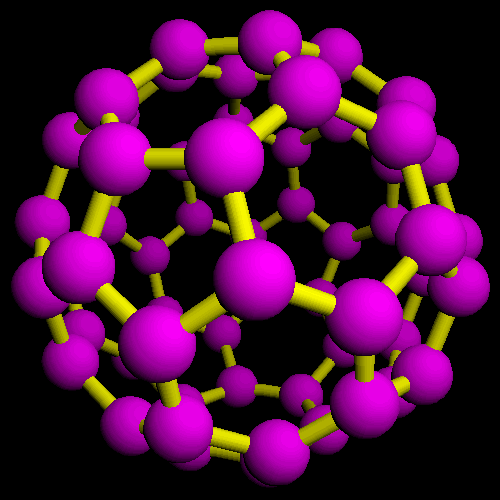
![]() Fullerene.
The full image is 48K
Fullerene.
The full image is 48K
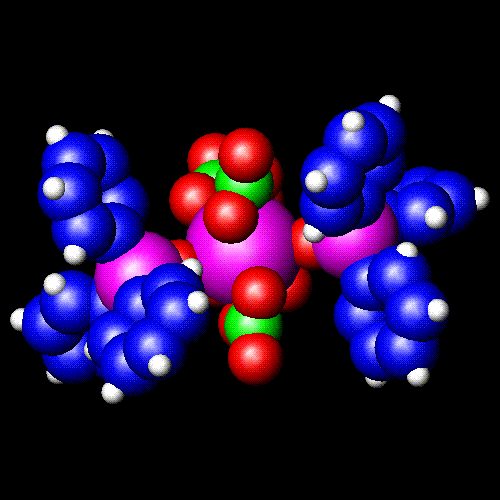
![]() Space-filling
(3D) The full image is 38K
Space-filling
(3D) The full image is 38K
![]() Epidote -
Hachures for atoms and polyhedra. The full image is 12K
Epidote -
Hachures for atoms and polyhedra. The full image is 12K
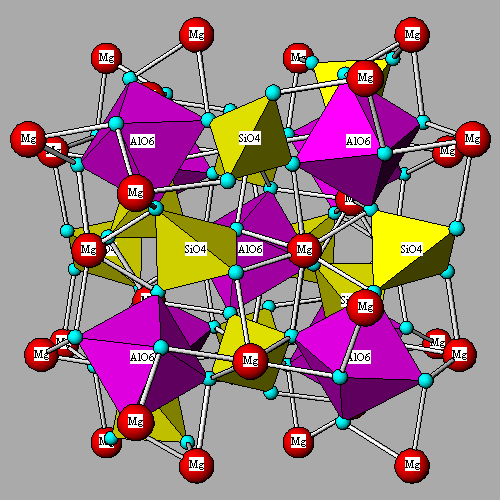
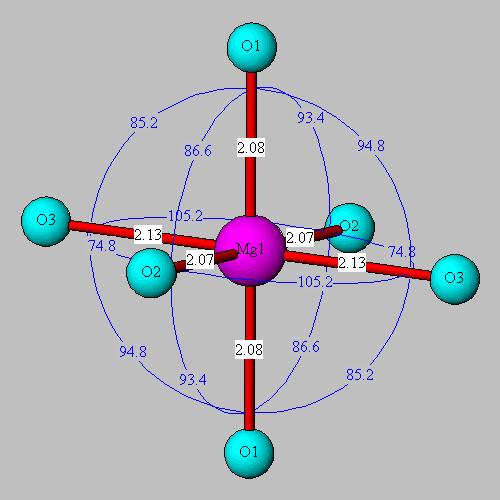
![]() MgO6
coordination in forsterite. The full image is 14K
MgO6
coordination in forsterite. The full image is 14K
![]() Albite twin
in low albite (NaAlSi3O8). The full image is 44K
Albite twin
in low albite (NaAlSi3O8). The full image is 44K

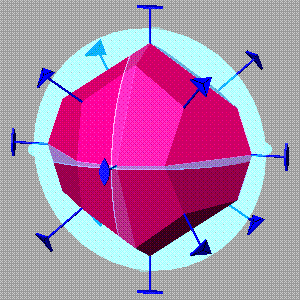
SHAPE for crystal morphology
SHAPE is a program for drawing the external morphology (faces) of crystals and quasi-crystals, and also for drawing sections of crystals. It will draw any single crystal and most twins and epitaxial intergrowths. You enter the crystal axis lengths and angles, the symbol for the crystal class, and one face of each form (symmetry equivalent set). The crystal can be rotated and scaled. A stereonet can be displayed with the crystal, or stereopairs can be drawn automatically. Screen display and hard copy of even complex, interpenetrating intergrowths may be made in final finished form, with no extraneous lines.
Quasi-crystals are crystal-like shapes with non-crystallographic symmetry such as icosahedral. SHAPE comes with an auxiliary program for generating the Cartesian symmetry matrices for any point symmetry group - the matrices for pentagonal and icosahedral groups are provided already.
Shape can draw sections through any crystal, at any orientation and depth. Moreover, sections can be draw sequentially at given time intervals for modeling crystal growth in section. The growth rates of individual forms can be constant with time, or can follow a general exponential growth-rate equation. This can be used to model the growth of twins, epitaxial intergrowths and even intergrowths of non-related crystals.
Symmetry-consistent striations may be drawn on any or all forms. Central distances, which determine face sizes, can be changed interactively with the mouse. Anaglyph (two-color stereopair) mode allows 3D viewing with standard red-green glasses. Faces can be labeled with indices or form letters.
Note: SHAPE Standard Edition (lacking quasi-crystals and sections) is no longer offered separately.
New for Version 6.0. The 3D display mode, which uses the OpenGL system software can give greater realism and allows shaded depiction of interpenetration twins and epitaxial intergrowths. In this mode it is possible to show symmetry elements, that is rotation axes and mirror planes. VRML (3D) files also can now show symmetry elements. The Donnay-Harker Morphology option can produce a list of forms with central distances dependent on the reticular density or X-ray d-spacing. The mouse cursor can be used to identify faces, resize faces (forms) or rotate the crystal. New forms can be added to truncate edges or corners selected with the mouse. A new Dialog Bar makes some common functions more accessible. Basic crystallographic data can now be imported from a number of external file types (see File Formats Supported).
New for Version 7.0 - 7.2. Additions include POV-Ray files and rotation movies.
New for Version 7.3. Now supports 3D stereoscopic viewing with either OpenGL quad-buffering or Microsoft Direct3D (DirectX).
New for Version 7.4. Supports writing files - StereoLithograph (.stl) and Wavefront (.obj) files - for direct output for 3D printing.
SHAPE for Windows V7.4. This is a 32-bit program which will run on Windows from XP on.
SHAPE for Macintosh V7.4. For Intel on OS 10.4 or later.
SHAPE for Linux/GTK V7.4. For Intel x86.
Crystal Libraries for SHAPE. The main library (700+ crystals) and supplementary library (200+ crystals), are based on the historical sets of wooden models designed in the 19th century by P. Groth and marketed by F. Krantz. Compiled by Ulrich Burchart.
Add SHAPE to your Shopping Cart
View some SHAPE images in raster format (may take some time to download).
![]() Cubic
multiface. The full image is 13K
Cubic
multiface. The full image is 13K
![]() Hourglass
zoning. The full image is 16K
Hourglass
zoning. The full image is 16K
![]() Icosahedron.
The full image is 7K
Icosahedron.
The full image is 7K
![]() Calcite
twin. The full image is 7K
Calcite
twin. The full image is 7K
![]() Phillipsite
twin. The full image is 16K
Phillipsite
twin. The full image is 16K
![]() Striated
pyrite. The full image is 10K
Striated
pyrite. The full image is 10K
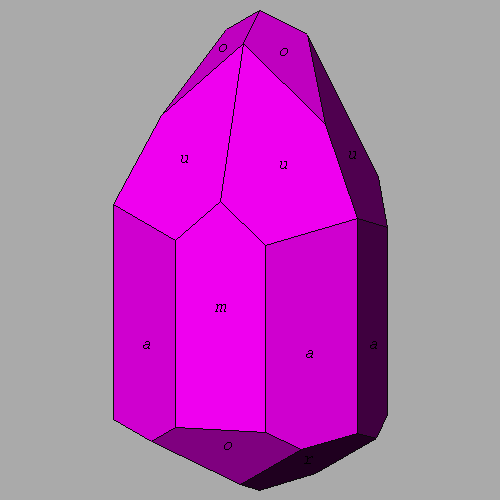
![]() Tourmaline.
The full image is 6K
Tourmaline.
The full image is 6K
VIBRATZ for normal-coordinate vibrational analysis
This program will do a complete normal-coordinate calculation on any molecule or crystal, using traditional valence force constants and/or Urey-Bradley force constants.
Input is: 1) the space group for crystals or polymers or the point group for molecules, selected from complete lists; 2) unit-cell parameters for crystals; 3) coordinates of the atoms, either a symmetry-unique set or all atoms; 4) specifications for force constants in terms of atom types (typically atomic numbers) and bond distance and angle limits.
The calculation is ultimately done in Cartesian coordinates, not internal coordinates, so there is no need to limit the number of force constants or to provide redundancy equations. Force-constant specifications can be easily transferred between structures. Forces may also be entered in the form of a Cartesian matrix, so that any (short-range) potential-energy function may be used.
Input data can be read from files with many crystallographic and molecular formats (see File Formats Supported).
Output is: 1) a complete list of frequencies of the fundamental vibrational modes (optical or kappa=0 modes in the case of crystals); 2) atomic motions in each mode; 3) potential-energy distribution; 4) plot of the structure with atomic motions; 5) synthetic infrared and Raman spectra.
VIBRATZ for Windows will run on any 32-bit Windows operating system. VIBRATZ for Mac will run on OS 8/9 or OS X (PPC and INTEL versions both supplied). VIBRATZ for Linux/GTK is for Intel x86.
VIBRATZ 2.0 has many additions, including: improved least-squares methods; Badger's rule variation of bond forces; new atom-by-atom options for force specification; potential-energy-distribution curves; z-matrix input for atomic positions and display of observed spectra.
New for V2.1 - 2.3. Multiple isotope configurations and conversion of Cartesian forces from GAUSSIAN or other programs to valence (internal) forces. Calculation of bond-length variation for EXAFS.
CRYSCON – general crystallographic conversion utility – Shareware.
Data can be imported from (and exported to) several popular crystallographic file formats (see File Formats Supported), through a "free-form" file input, or entered manually.
You can convert to a sub-cell or
super-cell or simply translate to a different origin or interchange
axes; or any combination of these. You can convert to
a sub- or super- symmetry group, or even a non-related space
group. When converting to a sub-cell or to higher symmetry the positions of
presumably coincident or superimposed atoms are averaged, and when converting to a super-cell or
lower symmetry the extra symmetry-unique atoms which may be required are
generated. In any case the result is a set
of
 symmetry-unique atoms for the target cell and symmetry, suitable for entry into
crystallographic software such as Fourier analysis, least-squares refinement or
structure drawing.
symmetry-unique atoms for the target cell and symmetry, suitable for entry into
crystallographic software such as Fourier analysis, least-squares refinement or
structure drawing.
Other features include computation of bond lengths and angles; simulation of powder diffractograms and precession photographs; transformation and reorientation of anisotropic temperature factors and magnetic or other vectors; and transformation of (hkl) index data, such as diffraction data.
New for version 2.0 is linkage to the FINDSYM program by H. T. Stokes and D. M. Hatch, which will determine the space-group symmetry of atoms in a unit cell.
Cryscon is shareware – the registration fee is $60.
Download CRYSCON for Macintosh
Add CRYSCON to your Shopping Cart
The following external formats are supported for input by ATOMS, SHAPE, VIBRATZ and CRYSCON (with some exceptions - for example non-crystallographic formats are not supported by SHAPE and CRYSCON). The various Shape Software programs can read files written by each other. CRYSCON can write crystallographic information in all the applicable formats. New formats are periodically added.
COD - Crystallography Open Database. Download files as CIF.
CCDC FDAT - files from the Cambridge Crystallographic Data Centre.
SHELX (.INS) - files from the program
system of G. Sheldrick.
CIF - Crystallographic Information Files. Includes mmCIF.
DBWS/LHPM - Rietveld input files.
ICSD Inorganic Crystal Structure Database
files.
Original - available
on-line through several
institutions
RETRIEVE and FINDIT
– recent and current CDROM
software formats
CAN/SND - files from
Canadian NRC
WWW
-
sample database on line
ORTEP - original or ORTEP II atom
information.
XTLVIEW - Drawing program.
PDB
- Protein Data Bank
files.
RIETAN - Rietveld program files.
GSAS - Los Alamos Lab system -
may include magnetic vectors.
AM MINERAL - Data files from the American
Mineralogist structure
data base.
FULLPROF - All-purpose refinement program
- may include magnetic
vectors.
IZA
Zeolites -
International Zeolite Association
files
Free-form - This allows input of atomic coordinates
and other information from almost any source.
GAUSSIAN - Atomic
coordinates will be read by ATOMS, and complete vibrational information by
VIBRATZ from a log file.
Ravel
Atoms archive
- This is for an XAFS program which is not the same as Shape Software ATOMS. The
.inp file format is not the same as the ATOMS free-form format.
VRML (Virtual Reality Modeling Language) files contain 3-dimensional information which is intended to recreate virtual worlds of all types. These files are a subset of the more extensive X3D standard. VRML files are platform-independent. Both ATOMS and SHAPE will write files of this type. The structures or crystals can be rotated and rescaled. In order to view such files, you need a 3-D plug-in for your Web Browser, or a stand-alone viewer. The CosmoPlayer and Cortona viewers are among the most popular plug-ins for Internet Browsers. See Web3D.org for the latest information on viewers.
View some VRML (3D) files. Requires a 3D plug-in and may take some time to download
![]() Forsterite fragment (ATOMS). The
full image is 19K
Forsterite fragment (ATOMS). The
full image is 19K
![]() Cavities in zeolite ZSM5 (ATOMS).
The full image is
264K
Cavities in zeolite ZSM5 (ATOMS).
The full image is
264K
![]() Calcite twin
(SHAPE). The full image is 38K
Calcite twin
(SHAPE). The full image is 38K
![]() Symmetry
elements in crystal class b3m (SHAPE). The full image is 37K
Symmetry
elements in crystal class b3m (SHAPE). The full image is 37K
Download Demos – Current
Releases
ATOMS for Windows V6.5 Demo (3.8M)
SHAPE for Windows V7.4
Demo (3.4M)
VIBRATZ for Windows
V2.3 Demo (2.9M)
CRYSCON for Windows
v2.0 (Shareware) (2.2M)
SHAPE and ATOMS for Windows require that Microsoft DirectX 9 or later be installed. If you get a message when you start up such as "d3dx9_42.dll not found" you should update to the latest DirectX. The best way to do this is directly from Microsoft:
An alternative, which is not recommended, is to install those files which are needed. As best as can be determined, at least the following are needed:
d3dx9_42.dll
d3d9.dll
d3d8thk.dll
These are usually installed in the \Windows\System32 directory. Installing dll's may require expert knowledge and incorrect installation may damage your system.
Demos for Macintosh - for Intel
All these programs are 32-bit and will run on Macintosh systems up to Mojave (OS 10.14), but will not run on Catalina (OS 10.15) which is 64-bit only. There are several ways to have both Catalina and older (32-bit) OS versions running on a single Macintosh - how to set this up is discussed on a separate page. Current Mac users may also be eligible for free or low-cost switch to the Windows version. Contact support@shapesoftware.com.
The Mac demo are in the form of .pkg installation files. After downloading and uncompressing, right-click on the package file and go ahead with the installation. By default the entire program folders are installed into the Applications folder on your hard disk but you can select another location.
ATOMS for Macintosh V6.5 Demo (4.7M)
SHAPE for
Macintosh V7.4 Demo (4.0M)
VIBRATZ
for Macintosh V2.3 Demo (3.2M)
CRYSCON for Macintosh V2.0
(Shareware) (1.8M)
Demos for Linux/GTK.
ATOMS for Linux/GTK
V6.5 Demo (13.8M)
SHAPE for Linux/GTK
V7.4 Demo (11.5M)
VIBRATZ for Linux/GTK V2.3 Demo (5.7M)
CRYSCON for
Linux/GTK V2.0 (Shareware) (4.6M)
Linux Demos all use the GTK environment (Intel x86 processor).
Detailed Linux installation instructions.
These Linux demos use fairly recent versions of the shared libraries. ATOMS and SHAPE have the Mesa OpenGL libraries statically linked so you do not need to have the dynamic versions of these libraries on your system. The Old Demos for Linux/GTK may be more satisfactory if you have an older Linux system, though they do require your system to have Mesa OpenGL with OSMesa.
As of 2003, all new versions of Shape Software programs are based on the wxWidgets cross-platform development framework, rather than the Microsoft Foundation Classes.
Bound instruction manuals have been discontinued. Each program has a manual in PDF form, which can be printed out. The Help system provided for each program contains all the material in the instructions.
 |
Prices and Ordering Information
Go to the SHOPPING CART for direct credit-card purchases. We now use PayPal, supporting VISA, MasterCard, American Express, Discover Card and Paypal accounts.
The following prices are for distribution directly by download or otherwise electronically as may be arranged. The instruction manual is included in electronic PDF form (see www.adobe.com for the free Adobe Acrobat reader).
Due to increasing postal rates and more complex customs rules we can no longer offer free shipping of CD-ROMs. Postal shipping of all the CD-ROMS in your order may be added as separate items at extra cost (see prices below)
For Linux versions, please download the demos and install before ordering.
ATOMS - Windows V6.5.....................................$380
ATOMS - Macintosh V6.5...................................$380
ATOMS - Linux V6.5.......................................$380
ATOMS - Library of 200+ Mineral Structures...............$100
SHAPE - Windows V7.4 ....................................$280
SHAPE - Linux V7.4 ......................................$280
SHAPE - Macintosh V7.4 ..................................$280
SHAPE - Libraries
1) Groth/Krantz Library of over 700 crystals.........$100
2) Groth/Krantz Supplementary Library
of over 200 crystals..............................$ 80
VIBRATZ - Windows V2.3...................................$380
VIBRATZ – Macintosh V2.3.................................$380
VIBRATZ - Linux V2.3.....................................$380
CRYSCON – Windows, Linux or Mac V2.0 - SHAREWARE
see above for download.................. .........$ 60
CD-ROM Distribution - USA Only...........................$ 10
CD-ROM Distribution - non-USA............................$ 40
Go to the SHOPPING CART for direct credit-card purchase.
For UPDATES, Call or email with your old version number(s) and/or purchase date. Then, go to the Updates category in the shopping cart and enter the amount you are given. If the amount is larger than the maximum, add items to get the correct total.
The above prices are for single-user licenses, allowing use by one person at a time (not restricted to a single computer). Departmental site licenses, for use by up to 12 persons at a time in a single department or institute, are double the above prices. Inquire about other types of license or pricing packages.We accept American Express, VISA, MasterCard or Discover Card (through PayPal - go to SHOPPING CART); company or institutional purchase orders; postal money orders: and personal checks (U.S.A. only). Non-U.S.A. customers please use a credit card (through the Paypal portal), or send an international money order, or a check or draft on a U.S. Bank (the bank routing number must be written on the check or draft).
SHAPE has been marketed since 1987, and ATOMS since 1989. Both are in use in universities and research laboratories throughout the world.
As always, technical support by email is free.
Shape
Software
521 Hidden Valley Road
Kingsport, TN 37663 USA
Email:
support@shapesoftware.com
10/10/2019
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}